Abstract
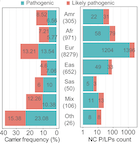
Background DNA sequencing is increasingly incorporated into the routine care of cancer patients, many of whom also carry inherited, moderate/high-penetrance variants associated with other diseases. Yet, the prevalence and consequence of such variants remain unclear. Methods We analyzed the germline genomes of 10,389 adult cancer cases in the TCGA cohort, identifying pathogenic/likely pathogenic variants in autosomal-dominant genes, autosomal-recessive genes, and 59 medically actionable genes curated by the American College of Molecular Genetics (i.e., the ACMG 59 genes). We also analyzed variant- and gene-level expression consequences in carriers. Results The affected genes exhibited varying pan-ancestry and population-specific patterns, and overall, the European population showed the highest frequency of pathogenic/likely pathogenic variants. We further identified genes showing expression consequence supporting variant functionality, including altered gene expression, allelic specific expression, and mis-splicing determined by a massively parallel splicing assay. Conclusions Our results demonstrate that expression-altering variants are found in a substantial fraction of cases and illustrate the yield of genomic risk assessments for a wide range of diseases across diverse populations.